Next-Generation Sequencing
January 15, 2022 | 4 min read

January 15, 2022 | 4 min read
The first venture into DNA sequencing was The Human Genome Project, a 13 year long, 3 billion dollar endeavor. The Human Genome Project was accomplished with the help of the first-generation Sanger sequencing. Since then, the need for accessible, faster, and efficient DNA sequencing methods has increased dramatically. This has resulted in the birth of Next-Generation Sequencing (NGS). NGS performs a process known as parallel sequencing. Millions of DNA sequences from a single sample are simultaneously sequenced, allowing the entire human genome to be sequenced within a day.
The methodology behind NGS involves three main steps:
Library Preparation
Sequencing and Imaging
Data Analysis
Library Preparation
This is the first step involved in NGS and includes building a library of nucleic acids and then amplifying it. DNA extracted from the cell or tissue is fragmented; these fragments are then converted into the library by ligating them with sequencing adapters containing specific sequences designed to interact with the NGS platform. Once built, these DNA libraries are clonally amplified and prepared for sequencing.
Sequencing and Imaging
DNA sequencing of whole chromosomes or whole genomes as well as targeted regions, such as an exome, can be carried out using NGS. The library's DNA fragments act as a template, off of which a new DNA fragment is created. The fragments formed are subjected to cycles of washing and flooding with known nucleotides in a sequential order. As these nucleotides get incorporated into the fragment, they are digitally recorded as a sequence.
Data Analysis
This includes both variant identification and annotation followed by visualization. Variant identification is a vital part of NGS data analysis. In this, sequence coverage is the main parameter, as identified mutations should be supported by several reads. Tools of variant identification are divided into four categories:
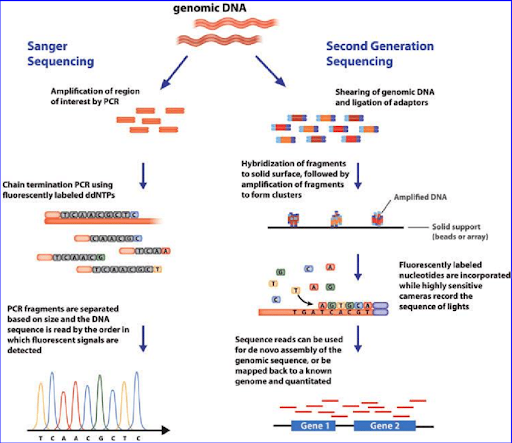
Figure 1. Source
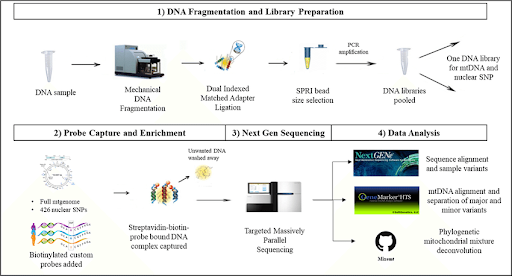
Figure 2. Source
May 16, 2021 | 3 min read

May 16, 2021 | 4 min read