Neural Progenitors Depleted by Zika Virus
Feb 02, 2021 | 3 min read

Feb 02, 2021 | 3 min read
Zika virus (ZIKV) belonging to the family Flaviviridae is an emerging mosquito-borne virus. This arbovirus infects the human skin with the help of a mosquito vector that can cause mild fever, rash, and joint pain for seven days or be asymptomatic in most cases. Moreover, ZIKV can cross the placental barrier to cause significant problems for pregnant women, including stillbirths as well as congenital disabilities. The virus is known to cause microcephaly, characterized by abnormally small brains, in new-borns.
Data suggests that ZIKV can cause microcephaly, but the study of the cells associated and the mechanism of infection, on pregnant women, is not easy. Therefore, one is reliant on in vitro methods. However, most of these methods do not account for the complexity of the brain. Hence, it is essential to use scalable, reproducible in vitro models capable of recapitulating complex neurodevelopmental events during early embryogenesis. The advances in embryonic stem cell (ESC) and induced pluripotent stem cell (iPSC) technology have opened up new avenues of disease in vitro modelling like the 3D organoid systems. The 3D organoid systems are able to differentiate, self-organize, and form distinct, complex and biologically relevant structures, thus making them ideal in vitro models of development, disease pathogenesis, and drug screening.
This paper ‘Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3.’ adopts the use of 3D organoid systems to investigate the mechanisms by which ZIKV induces microcephaly and other neurological disorders. They hypothesized that ZIKV activates the TLR3-mediated innate immune response, leading to dysregulation of a network of genes involved in neurogenesis, axon guidance, apoptosis, and differentiation.
Here, human ESC (hESC)-derived cerebral organoids were used to investigate the role of ZIKV in microcephaly. The cerebral organoids generated from hESCs mimic the developing foetal brain. Then, to determine the effect of ZIKV on foetal brain tissue in vitro, human cerebral organoids were infected with a ZIKV strain and growth of the cerebral organoids were tracked for five days post-infection and then compared with a healthy cerebral organoid. It was observed that the healthy organoids showed an average of 22.6% increase in growth. In comparison, ZIKV-infected organoids significantly decreased by 16%, therefore resulting in a net 45.9% difference in size on average.
Moreover, a strong co-localization of ZIKVE (a marker for ZIKV) in NESTIN (a marker for neural progenitors cells) -positive cell populations compared to NESTIN-negative cells was observed, indicating that ZIKV infects NPCs in organoid models.
Previous studies showed that ZIKV activates TLR3, and the upregulation of TLR3 was observed in cerebral organoids and mouse neurospheres (culture system of neural stem cells) after ZIKV infection by RT-qPCR analysis.
To study the link between ZIKV-mediated TLR3 activation and dysregulation of neurogenesis and apoptosis, they investigated the effect of TLR3 agonist (poly(I:C)) and a high-affinity inhibitor of TLR3 inhibitor on mouse neurospheres and human organoids. To determine the effect of TLR3 activation, neurospheres challenged with poly(I:C) exhibited a statistically significant decrease in overall neurosphere size relative to mock-treated organoids. Further, when neurospheres were inoculated with ZIKV with TLR3-competitive inhibitor. A significant difference between ZIKV-treated neurospheres with and without TLR3 inhibitor was observed. Now, to prove their hypothesis, the cerebral organoids were treated with a TLR3-competitive inhibitor in the presence of ZIKV. As expected, the TLR3 competitive inhibitor attenuated the severe ZIKV-mediated apoptosis and organoid shrinkage that strongly suggested that TLR3 may play a crucial role in the ZIKV-associated phenotype.
Further to study the role of TLR3 activation, the differentially expressed genes involved in the organoid formation and TLR-regulated genes were obtained to identify the common pathways that were activated upon ZIKV infection. And it was found that ZIKV infection and poly(I:C) treatment of organoids reduced NTN1(axon guidance, apoptosis, neural cell death, and cellular reprogramming) and EPHB2(radial migration, proliferation, and cell fate of NPC in the subventricular zone) expression. Additionally, TLR3 competitive inhibitor reversed the downregulation of NTN1 and EPHB2 on ZIKV infection. This concluded that ZIKV perturbs a TLR3-regulated network controlling neurogenesis and apoptotic pathways.
Using robust, reproducible and scalable human cerebral organoid models, this paper presents evidence that TLR3 activation triggers apoptosis and attenuates neurogenesis, that may strongly contribute to ZIKV-mediated microcephaly phenotype.
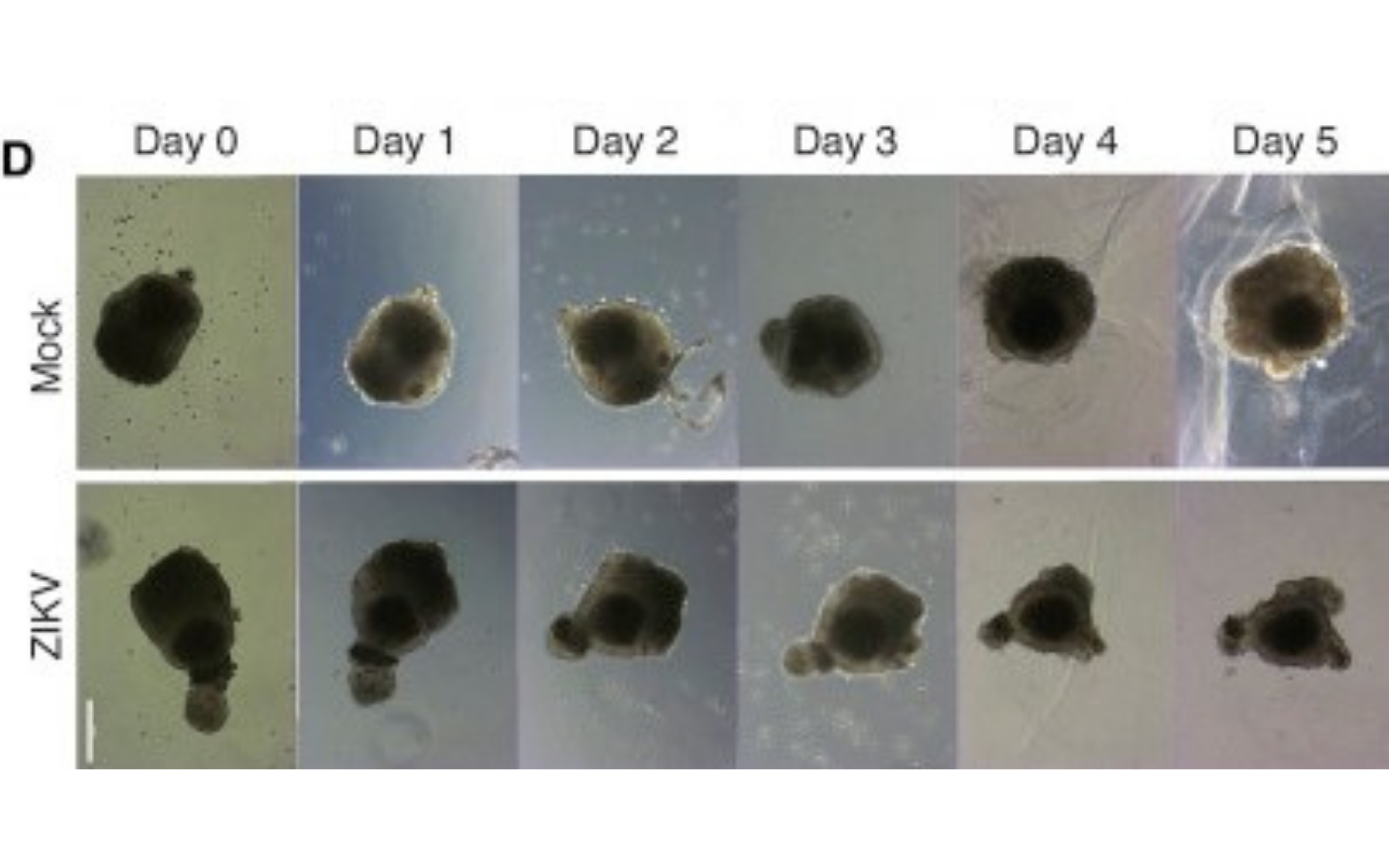
Figure 1. Representative bright-field images of individual human cerebral organoids treated with ZIKV over time. Scale bar represents 250 μm.
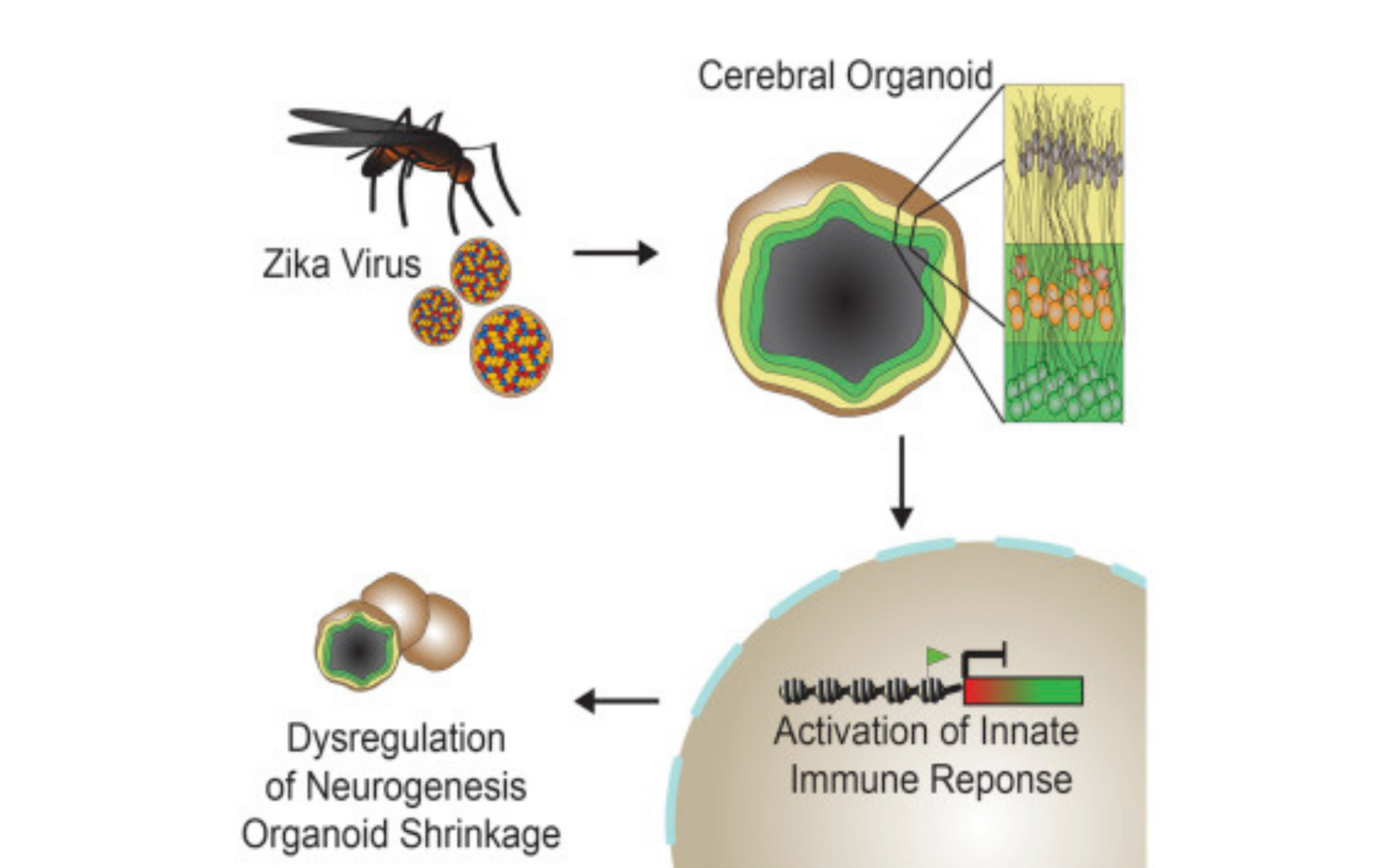
Figure 2. Graphical Abstract

Feb 02, 2021 | 2 min read

Oct 31, 2020 | 1 min read